



scanpy对单细胞测序数据的聚类分析
scanpy对scRNA-seq数据的聚类分析
R在读取和处理数据的过程中会将所有的变量和占用都储存在RAM当中,这样一来,对于海量的单细胞RNA-seq数据(尤其是超过250k的细胞量),即使在服务器当中运行,Seurat、metacell、monocle这一类的R包的使用还是会产生内存不足的问题。
但是一个基于python的单细胞基因表达分析包scanpy,能够很好地在仅4G内存的小破机上分析378k的细胞,并且功能丰富程度不亚于Seurat。它包含了数据预处理、可视化、聚类、伪时间分析和轨迹推断、差异表达分析。根据官网描述,scanpy能够有效处理超过 1,000,000个细胞的数据集。
Scanpy – Single-Cell Analysis in Python:https://scanpy.readthedocs.io/en/latest/
安装与数据下载
安装好python3之后,终端运行:
pip install scanpy
若安装过程出现问题,请参考:
https://scanpy.readthedocs.io/en/latest/installation.html
骨髓单细胞转录组测序数据下载地址:
https://preview.data.humancellatlas.org
Step0, 读取数据
运行python
import numpy as np
import pandas as pd
import scanpy as sc
# 可以直接读取10Xgenomics的.h5格式数据
adata = sc.read_10x_h5("/Users/shinianyike/Desktop/ica_bone_marrow_h5.h5"
, genome=None, gex_only=True)
adata.var_names_make_unique()
查看数据:
adata
AnnData object with n_obs × n_vars = 378000 × 33694
var: 'gene_ids'
共378000个细胞,33694个基因。
Step1, 数据预处理
这一步目的将数据进行筛选和过滤,一些测序质量差的数据会让后续的分析产生噪音和干扰,因此我们需要将它们去除。
展示在所有的细胞当中表达占比最高的20个基因。
sc.pl.highest_expr_genes(adata, n_top=20)
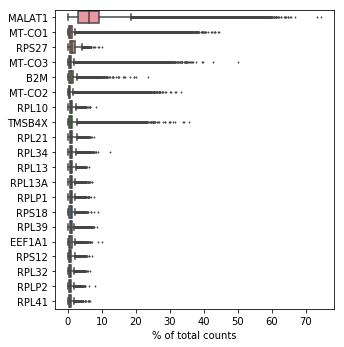
表达水平前20的基因.png
基础过滤:
去除表达基因200以下的细胞;
去除在3个细胞以下表达的基因。
sc.pp.filter_cells(adata, min_genes=200) # 去除表达基因200以下的细胞
sc.pp.filter_genes(adata, min_cells=3) # 去除在3个细胞以下表达的基因
adata
AnnData object with n_obs × n_vars = 335618 × 24888
obs: 'n_genes'
var: 'gene_ids', 'n_cells'
共335618个细胞,24888个基因。(可以发现细胞跟基因数量都减少了)
质量控制:
在测序后,有很大比例是低质量的细胞,可能是因为细胞破碎造成的胞质RNA流失,由于线粒体比单个的转录分子要大得多,不容易在破碎的细胞膜中泄漏出去,这样一来就导致测序结果显示线粒体基因的比例在细胞内占比异常高。这一步质量控制的目的就是将这些低质量的细胞去除掉。
计算线粒体基因占所有基因的比例:
mito_genes = adata.var_names.str.startswith('MT-')
# for each cell compute fraction of counts in mito genes vs. all genes
# the `.A1` is only necessary as X is sparse (to transform to a dense array after summing)
adata.obs['percent_mito'] = np.sum(
adata[:, mito_genes].X, axis=1).A1 / np.sum(adata.X, axis=1).A1
# add the total counts per cell as observations-annotation to adata
adata.obs['n_counts'] = adata.X.sum(axis=1).A1
作小提琴图,查看线粒体基因占比分布:
sc.pl.violin(adata, ['n_genes', 'n_counts', 'percent_mito'],
jitter=0.4, multi_panel=True)
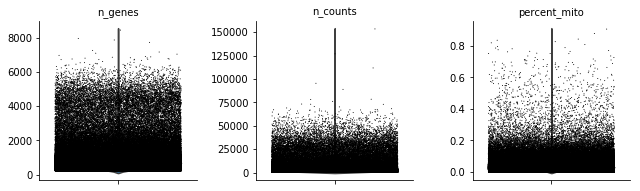
细胞表达的基因种数、基因数、线粒体基因占比
由于数据点实在太多,小提琴已被数据点覆盖,无法显示出来。
这里还可以做一个散点图,也可以直观地显示出一些异常分布的数据点。
sc.pl.scatter(adata, x='n_counts', y='percent_mito')
sc.pl.scatter(adata, x='n_counts', y='n_genes')


adata
AnnData object with n_obs × n_vars = 335618 × 24888
obs: 'n_genes', 'percent_mito', 'n_counts'
var: 'gene_ids', 'n_cells'
335618个细胞,24888个基因;
下面这一步进行真正的过滤,根据上面做的图,去除基因数目过多(大于等于4000)和线粒体基因占比过多(大于等于0.3)的细胞:
adata = adata[adata.obs['n_genes'] < 4000, :]
adata = adata[adata.obs['percent_mito'] < 0.3, :]
过滤后查看剩下多少细胞和基因。
adata
View of AnnData object with n_obs × n_vars = 328435 × 24888
obs: 'n_genes', 'percent_mito', 'n_counts'
var: 'gene_ids', 'n_cells'
328435个细胞,24888个基因。
数据标准化
在绘图之前,还要进行一步数据标准化,将表达量用对数计算一遍,这样有利于绘图和展示。
sc.pp.normalize_per_cell(adata, counts_per_cell_after=1e4)
sc.pp.log1p(adata)
adata.raw = adata # 储存标准化后的AnnaData Object
识别差异表达基因
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
sc.pl.highly_variable_genes(adata)
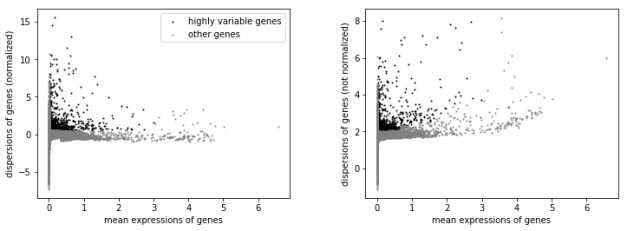
将保守的基因去除,留下差异表达的基因用于后续分析。
adata = adata[:, adata.var['highly_variable']]
adata
View of AnnData object with n_obs × n_vars = 328435 × 1372
obs: 'n_genes', 'percent_mito', 'n_counts'
var: 'gene_ids', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
328435个细胞,1372个基因。
回归每个细胞总计数和线粒体基因表达百分比的影响。将数据放缩到方差为1。单细胞数据集可能包含“不感兴趣”的变异来源。这不仅包括技术噪音,还包 括批次效应,甚至包括生物变异来源(细胞周期阶段)。正如(Buettner, et al NBT,2015)中所建议的那样,从分析中回归这些信号可以改善下游维数减少和聚类。
这一步高内存需求预警,建议清理电脑缓存,关闭后台不使用的应用。
sc.pp.regress_out(adata, ['n_counts', 'percent_mito'])
/Users/shinianyike/anaconda3/lib/python3.6/site-packages/statsmodels/compat/pandas.py:56: FutureWarning: The pandas.core.datetools module is deprecated and will be removed in a future version. Please use the pandas.tseries module instead.
from pandas.core import datetools
Scale each gene to unit variance. Clip values exceeding standard deviation 10.
sc.pp.scale(adata, max_value=10)
Step2, 主成分分析
主成分分析是一种将数据降维的分析方法,是考察多个变量间相关性一种多元统计方法,研究如何通过少数几个主成分来揭示多个变量间的内部结构,即从原 始变量中导出少数几个主成分,使它们尽可能多地保留原始变量的信息,且彼此间互不相关.通常数学上的处理就是将原来P个指标作线性组合,作为新的综合指 标。
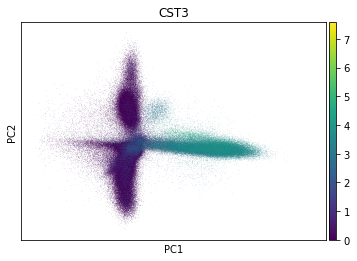
sc.tl.pca(adata, svd_solver='arpack') # PCA分析
sc.pl.pca(adata, color='CST3') #绘图
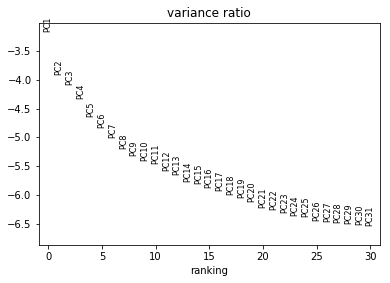
作碎石图,观测主成分的质量。这个图用于选择后续应该使用多少个PC,用于计算细胞间的相邻距离。
sc.pl.pca_variance_ratio(adata, log=True)
在这里先将数据保存,便于后续操作的更改。
adata.write("pca_results.h5ad")
Step3, 聚类分析
计算细胞间的距离:
这里的参数就先按照默认值设定:
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
参数说明:
n_neighbors指的是每个点的邻近点的数量,据评论区@小光amateur 所说neighbors的个数越多,聚类数会越少。
pc的数量依赖于上面所做的碎石图,一般是选在拐点处的的主成分,只需要一个粗略值,不同的pc数量所产生的聚类形状也不同。我后来更改为PC=16,效果比下图要好一些。
将距离嵌入图中:
sc.tl.umap(adata)
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'])

sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'], use_raw=False)

聚类
sc.tl.louvain(adata)
sc.pl.umap(adata, color=['louvain'])

output_51_0.png
这里得到了29类细胞,每个颜色代表一种。
将数据保存。
adata.write("umap.h5ad")
寻找marker基因
marker基因通常是细胞表面抗原,用于定义出该细胞的细胞类型。
为了定义每个簇属于什么细胞,根据基因的差异表达水平,将每个簇排名前25的基因导出。
sc.tl.rank_genes_groups(adata, 'louvain', method='wilcoxon')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)

下一步的工作是找出每一个簇的marker基因对应的细胞类型,这主要依靠一些数据库或生物学的相关背景知识。
想问用marker基因注释细胞类型除了SingleR包还有什么办法吗?这个包一直都装不上
感觉SingleR是最好的自动注释包,装不上的话可以提问作者,或看看其他人的提问,也许有和你一样的人
可以问你一个问题么 为什么我安装scanpy一直安装失败 提示can not uninstall llvmlite 请问 你遇到这个问题了么 我用的python3.6.5
没有遇到,建议去GitHub上看看其他人有没有跟你一样的问题
博主,你在跑这个流程的时候用的是linux系统还是windows?我用windows10系统测试的时候,本子16G内存,还是不够用~
我用的是苹果,才4G内存
大神,sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)中的neighbor和pc个数设置的莫名其妙的,这样设置有什么依据呢?另外,聚类出那么多的cluster,难道不会增加cell marker寻找的难度吗。而且不会出现很多重复的细胞类型吗?
这个我是按照scanpy给的默认值设的,pc选一个粗略值就行,一般在上面那个pc碎石图的拐点处,我后来改成了pc = 16,得到的聚类效果比这个好一些。至于neighbors就比较复杂,是临近数据点的数量,可以设2-100之间某个数,我也不知道调成其他样子会怎么样,你可以调下试试hhh;
至于你说的出现重复细胞类型,如果对于我们常说的大类来说,确实会这样,而且找细胞类型真的很麻烦!!!!但另一方面说,如果都是T细胞,为什么会被聚成两个不同的cluster?是否是不同的细胞亚型或者甚至是某一种新的细胞呢?所以聚类聚出来的cluster数量需要多少就看研究目的了。
嗯嗯,万分感谢,试了一下,那个neighbors的个数越多,聚类数会越少,所以这个值对结果影响真的很大。这个软件真的很玄学啊

-
- 2021-03-31
- 2020-07-31
- 2019-09-16
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29
-
- 2021-03-31
- 2020-07-31
- 2019-09-16
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29


-
- 2021-03-31
- 2020-07-31
- 2019-09-16
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29

