



用R包Seurat进行QC、PCA分析与t-SNE聚类
用R包Seurat进行QC、PCA分析与t-SNE聚类
版本信息:
Seurat v2.0不是3.0!现在Seurat更新了3.0版本,下载也是默认的3.0,这篇记录只适用于用2.0的
内容
1. 将Cellranger中的基因表达矩阵filtered_gene_bc_matrices用于分析
2. 进行质量控制(QC),以删除异常细胞
3. 标准化与归一化,消除技术噪音与批次效应
4. 主成分分析(PCA)与挑选
5. t-SNE聚类
参考网站:https://satijalab.org/seurat/pbmc3k_tutorial.html
(注意!!!现在这个网站会自动跳转到3.0版本)
Seurat的安装:R中运行install.packages("Seurat")
上次结果:
经过Cellranger的数据整理之后,得到:
· Filtered gene-barcode matrices MEX: /outs/filtered_gene_bc_matrices
此输出结果应为基因-细胞的表达矩阵,用Seurat包进行后续分析。
Seurat是一种R包,设计用于QC,分析和探索单细胞RNA-seq数据。 Seurat旨在使用户能够从单细胞转录组测量中识别和解释异质性来源,并整合不同类型的单细胞数据。
运行R,并且加载这两个包
library(Seurat)
library(dplyr)
读取数据
spleen.data <- Read10X(data.dir = '/GRCh38/')
dim(spleen.data)
[1] 33694 1960
原始数据的基因数为33694,细胞数为1960.
比较普通与疏松矩阵的内存使用:
> dense.size <- object.size(x = as.matrix(x = spleen.data))
> dense.size
530488272 bytes
#转化为疏松矩阵,查看大小
> sparse.size <- object.size(x = spleen.data)
> sparse.size
45955656 bytes
> dense.size/sparse.size
11.5 bytes
初始化Seurat对象:
命令CreateSeuratObject
输入数据spleen.data
留下所有在>=3个细胞中表达的基因min.cells = 3;
留下所有检测到>=200个基因的细胞min.genes = 200。
(为了除去一些质量差的细胞)
spleen <- CreateSeuratObject(raw.data = spleen.data, min.cells = 3, min.genes = 200, project = "10X_spleen")
spleen
An object of class seurat in project 10X_spleen
15655 genes across 1959 samples.
剩下15655 基因和 1959 个细胞
质量控制
以下步骤包括Seurat中scRNA-seq数据的标准预处理工作流程。这些代表了Seurat对象的创建,基于QC指标的细胞选择和过滤,数据标准化和缩放,以及高度可变基因的检测。
mito.genes <- grep(pattern = "^MT-", x = rownames(x = spleen@data), value = TRUE)
percent.mito <- Matrix::colSums(spleen@raw.data[mito.genes, ])/Matrix::colSums(spleen@raw.data)
spleen <- AddMetaData(object = spleen, metadata = percent.mito, col.name = "percent.mito")
VlnPlot(object = spleen, features.plot = c("nGene", "nUMI", "percent.mito"), nCol = 3)
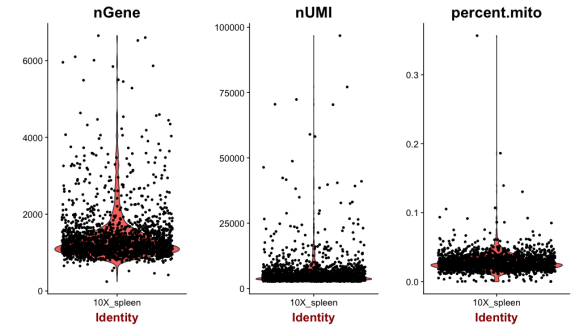
VlnPlot_of_spleen.png
> par(mfrow = c(1, 2))
> GenePlot(object = spleen, gene1 = "nUMI", gene2 = "percent.mito")
> GenePlot(object = spleen, gene1 = "nUMI", gene2 = "nGene")
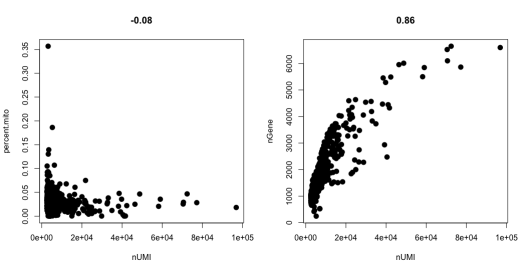
GenePlot_of_spleen.png
过滤细胞,根据上面的两幅图,去除异常值,这里选择基因数从300-5000,线粒体基因占比大于0.1的细胞。(主要看小提琴图1和图3)
spleen <- FilterCells(spleen, subset.names = c("nGene", "percent.mito"), low.thresholds = c(300, -Inf), high.thresholds = c(5000,0.10))
查看过滤掉剩下多少细胞:
spleen
An object of class seurat in project 10X_spleen
15655 genes across 1940 samples.
剩下15655个基因,1940个细胞。
数据标准化
加个log:
spleen <- NormalizeData(object=spleen, normalization.method = "LogNormalize", scale.factor = 10000)
Performing log-normalization
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
spleen <- FindVariableGenes(object = spleen, mean.function = ExpMean, dispersion.function = LogVMR, x.low.cutoff = 0.0125, x.high.cutoff = 3, y.cutoff = 0.5)
Calculating gene means
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
Calculating gene variance to mean ratios
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
TEXT_SHOW_BACKTRACE environmental variable.
> length(x=spleen@var.genes)
[1] 1829
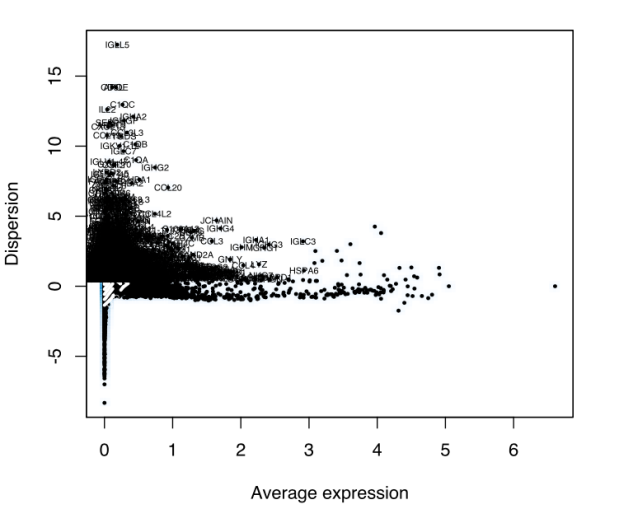
高度变异基因.png
缩放数据并删除不需要的变体来源
您的单细胞数据集可能包含“不感兴趣”的变异来源。这不仅包括技术噪音,还包括批次效应, 甚至包括生物变异来源(细胞周期阶段)。正如(Buettner, et al NBT,2015)中所建议的那样,从分析中回归这些信号可以改善下游维数减少和聚类。为了减轻这些信号的影响,Seurat构建线性模型以基于用户定义 的变量预测基因表达。这些模型的缩放得分残差存储在Scale.data槽中,用于降维和聚类。
我们可以消除由批次(如果适用)驱动的基因表达中的细胞 - 细胞变异,细胞比对率(由Drop-seq数据的Drop-seq工具提供),检测到的分子数量和线粒体基因表达。对于循环细胞,我们还可以学习“细胞周 期”评分(参见此处的示例)并对其进行回归。在这个有丝分裂后血细胞的简单例子中,我们回归了每个细胞检测到的分子数量以及线粒体基因含量百分比。
spleen <-ScaleData(spleen, vars.to.regress = c("nUMI","percent.mito"))
Regressing out: nUMI, percent.mito
|=========================================================================================| 100%
Time Elapsed: 18.0711550712585 secs
Scaling data matrix
|=========================================================================================| 100%
PCA分析
主成分分析是什么?
主成分分析,是考察多个变量间相关性一种多元统计方法,研究如何通过少数几个主成分来揭示多个变量间的内部结构,即从原始变量中导出少数几个主成 分,使它们尽可能多地保留原始变量的信息,且彼此间互不相关.通常数学上的处理就是将原来P个指标作线性组合,作为新的综合指标。
将数据集降维,利用低阶的变量去反应整体的结果。
spleen <- RunPCA(spleen, pc.genes = spleen@var.genes, do.print = TRUE, pcs.print = 1:5, genes.print = 5)
[1] "PC1"
[1] "CD69" "CD79A" "TRAC" "CD3D" "MS4A1"
[1] ""
[1] "FCN1" "LYZ" "SERPINA1" "CSTA" "RP11-1143G9.4"
[1] ""
[1] ""
[1] "PC2"
[1] "CD79A" "MS4A1" "VPREB3" "CD79B" "HLA-DQB1"
[1] ""
[1] "NKG7" "CST7" "GZMA" "CD7" "CCL5"
[1] ""
[1] ""
[1] "PC3"
[1] "TRDC" "KLRF1" "MS4A1" "CD79B" "IRF8"
[1] ""
[1] "IL7R" "TRAC" "CD3D" "CD2" "CD3G"
[1] ""
[1] ""
[1] "PC4"
[1] "GIMAP7" "GZMB" "FGFBP2" "SPON2" "PRF1"
[1] ""
[1] "BAG3" "HSPD1" "FKBP4" "DNAJA1" "ZFAND2A"
[1] ""
[1] ""
[1] "PC5"
[1] "UBE2C" "TYMS" "MKI67" "TOP2A" "AURKB"
[1] ""
[1] "FCGR3A" "FGFBP2" "SPON2" "GNLY" "GZMB"
[1] ""
[1] ""
PCElbowPlot(spleen)
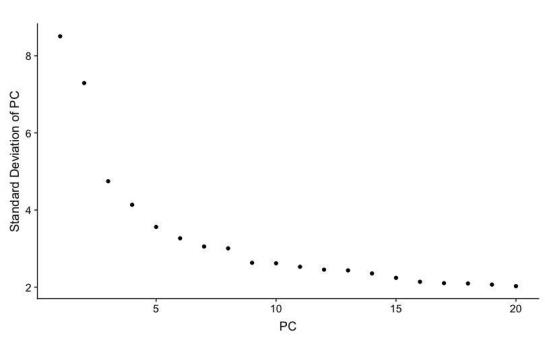
碎石图.jpeg
选择了前10个PC成分
spleen <- FindClusters(spleen, reduction.type = "pca", dims.use = 1:10, resolution = 0.6, print.output = 0, save.SNN = TRUE)
PrintFindClustersParams(spleen)
Parameters used in latest FindClusters calculation run on: 2018-10-01 21:59:55
=============================================================================
Resolution: 0.6
-----------------------------------------------------------------------------
Modularity Function Algorithm n.start n.iter
1 1 100 10
-----------------------------------------------------------------------------
Reduction used k.param prune.SNN
pca 30 0.0667
-----------------------------------------------------------------------------
Dims used in calculation
=============================================================================
1 2 3 4 5 6 7 8 9 10
细胞聚类
spleen <- RunTSNE(spleen, dims.use = 1:10, do.fast= TRUE)
TSNEPlot(spleen)
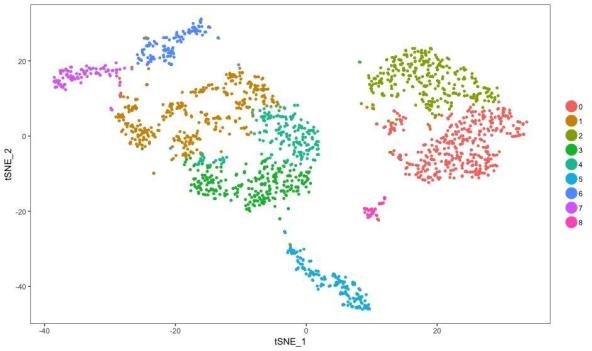
TSNE.jpeg
> saveRDS(spleen, file = "/spleen_1.rds")
将R变量保存,利于后续的分析。
一些补充:
过滤低质量细胞:
在 scRNA-seq 分析中,有些细胞质量比较低,比如细胞处于凋亡状态,细胞中 RNA 发生降解等,这些细胞的存在会影响分析,因此我们第一步需要对细胞进行过滤。主要可分为三类:
①利用细胞检测到的基因数或者是 reads 比对率来判断技术噪音。
但不管是基因检测数目还是比对率都跟实验方法有很大相关性。 如果比对率太低,表明 RNA 可能发生了降解,或者文库有污染或者细胞裂解不完全。
②如果实验中加入了 spike-ins(本实验没有),可以通过计算比对到内源性 RNA 和外源性 RNA(spike-ins)的 reads 比例来过滤低质量细胞。
比值偏低表明细胞中的 RNA 数量较低,细胞可丢弃。但是也需要注意其实当细胞状态不一样,比如处于不同细胞周期时,细胞的 RNA 数量是具有很大差异的。不过我们依然认为在一大群细胞中,spike-ins比例特别高的细胞在很大概率上应该被排除在外。软件 SinQC (Single-cell RNA-seq Quality Control)可以根据比对率和检测到的基因数来过滤细胞。
③根据整体的基因表达谱来定义技术噪音。
比如对细胞进行聚类分析,PCA 分析等,将 outlier 细胞删除,或者细胞表达中位值低于某一设定阈值时将该细胞过滤掉。当然这种方法也存在误删具有真正生物学差异的细胞,因此在删除细胞时需要小心,可与上述另外两种方法连用。
如果你的数据量过大,使用Seurat时内存不足,请看
《海量scRNA-seq数据的质量控制、PCA、聚类》
(你好,我想问你下你,我跑“pbmc <- CreateSeuratObject(raw.data = pbmc.data, min.cells = 3, min.genes = 200, project = "10X_PBMC")”这一步时候出现以下错误-------Error in CreateSeuratObject(raw.data = pbmc.data, min.cells = 3, min.genes = 200, :
unused arguments (raw.data = pbmc.data, min.genes = 200),实在看不明白,请教你一下~谢谢,还有(你总结的ggplot笔记特别棒)
谢谢!应该是版本的问题,我写这篇记录的时候还只有旧版,现在你用的版本如果是3.0的话所用的参数就不一样了
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
)

-
- 2021-03-31
- 2020-07-31
- 2019-09-16
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29
-
- 2021-03-31
- 2020-07-31
- 2019-09-16
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29


-
- 2021-03-31
- 2020-07-31
- 2019-09-16
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29
- 2019-08-29

